
OpenMS – from mass spectra to knowledge
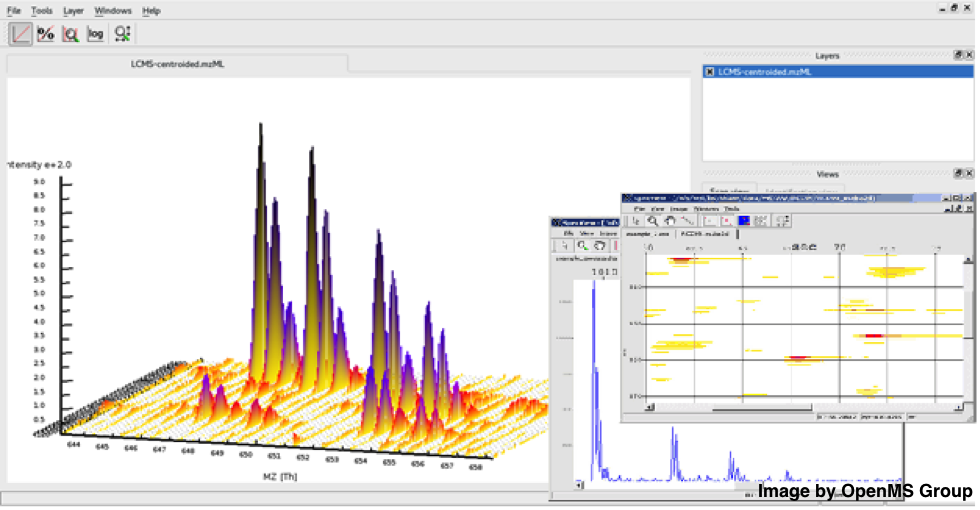 Figure 1: Inspect your raw mass spectra and run tools from within the OpenMS visualisation tool TOPPViewFigure 1: Inspect your raw mass spectra and run tools
Figure 1: Inspect your raw mass spectra and run tools from within the OpenMS visualisation tool TOPPViewFigure 1: Inspect your raw mass spectra and run toolsfrom within the OpenMS visualisation tool TOPPView
By Timo Sachsenberg and Oliver Kohlbacher, University of Tübingen
This article is part of our series A day in the software life, in which researchers from all disciplines discuss the tools that make their research possible.
High-throughput mass spectrometry has become a versatile technique to tackle a large range of questions in the life sciences. Being able to quantify diverse classes of biomolecules opens the way for improved disease diagnostics, elucidation of molecular structure and investigation of cellular pathways. In an interplay with other open-source software, OpenMS enables powerful workflows to transform biological data into meaningful knowledge.
In recent years, mass spectrometry has gained significant attention in the life sciences. The mass spectrometer determines the mass and abundance of ionised molecules like proteins, metabolites or lipids and stores them as so-called mass spectra. Speed and mass resolution have been steadily increasing to the point where we can, for instance, identify and quantify thousands of proteins in a biological sample.
With the increased speed and sensitivity of modern machines comes the problem of dealing with a vast amount of data. But not only the data deluge poses significant problems— every new experimental setup needed to tackle a scientific question requires custom data processing.
While commercial and open software solutions exist for most standard analysis tasks, the world appears different for many scientists. Experimentalists often develop novel methods or need to analyse huge data sets on computer clusters.
We run a computational mass spectrometry lab composed of bioinformaticians that regularly deal with the fact that no existing software can perform the required analysis tasks or is too restricted to run on large computer infrastructures. To cope with this problem, we started the development of OpenMS as a software library for C++ developers who want to implement novel algorithms. Over more than ten years, OpenMS has become one of the leading bioinformatics solutions in the field with an active user and developer community. In addition to being a software library, OpenMS provides more than 185 processing tools. These tools were built based on the software library and allow constructing powerful analysis workflows with ease. Common analysis tasks, typically carried out by these tools, comprise signal processing, protein sequencing, identification and quantification, among others. A common interface and use of open, standardised data formats enable chaining of tools into complex, custom-tailored analysis workflows.
Some recent examples of the use of OpenMS workflows demonstrate how important scientific questions can be tackled with a minimum of development effort. OpenMS users have build workflows and tools for the analysis of protein abundances in human blood plasma to study the effect and assess the toxicity of nanomaterial, the investigation of protein degradation by human enzymes, the study of protein-RNA interactions, and the refinement of protein-coding gene annotations in the human genome. In all of these cases, maximum reusability has been achieved by using existing OpenMS tools or by developing missing tools using the OpenMS library.
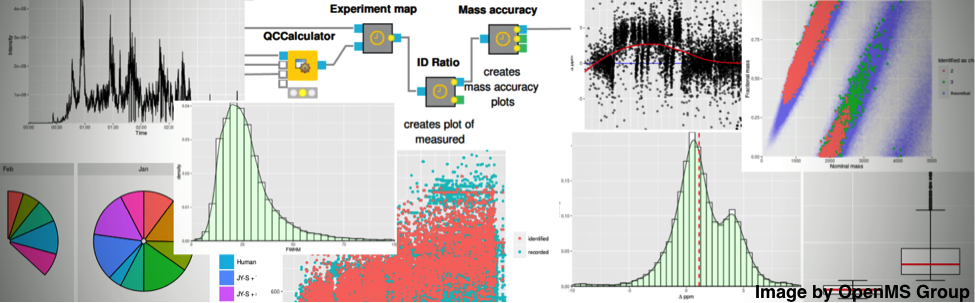 Figure 2: Integration of OpenMS into KNIME enables statistical downstream processing and visual analytics.Figure 2: Integration of OpenMS into KNIME enables statistical downstream processing and visual analytics.
Figure 2: Integration of OpenMS into KNIME enables statistical downstream processing and visual analytics.Figure 2: Integration of OpenMS into KNIME enables statistical downstream processing and visual analytics.OpenMS greatly benefits from a range of open-source projects by wrapping their functionality for file conversion or protein identification. To package these third-party tools into a coherent solution, we deploy OpenMS on all major platforms (Linux, macOS, Windows) as both self-contained, stand-alone installers for command line execution—e.g. on a cluster environment—and plugins for integration into workflow systems. A powerful and plugin-extendable deployment platform is KNIME (KoNstanz Information MinEr). KNIME is an open-source, industry-supported integration workflow system with wide adoption in pharmaceutical research.
In addition to the OpenMS plugin, hundreds of third-party plugins provide analysis tools for advanced statistics, integration of scripting languages, machine learning, database connectors or automated internet queries, among others. This enables us to perform mass spectrometry data processing as well as downstream statistical data analysis in a single workflow. We also benefit from KNIME as it allows storing the full analysis workflow including all parameters and scripts. That is, other projects can reuse parts of the workflow, and, even more importantly, the complete analysis task becomes reproducible—a central aspect of scientific research that many fields don’t give it the attention it deserves.
Apart from development, we provide free consulting and regular training events to users from different backgrounds. Ultimately, our aim is to generate new knowledge by getting novel methods promptly to our users.
If you would like to find out more, please visit the OpenMS website.